
Haplotag linked-reads
A low-cost linked-read technology for the masses
 Azwad Iqbal
Azwad Iqbal  Pavel Dimens
Pavel Dimens What is linked-read sequencing?
Next-generation sequencing has rapidly expanded genetics and genomics research. Short-read sequencing remains the most cost-effective and high-throughput option, while long-read technologies provide advantages like better genome assemblies, longer haplotypes, and improved detection of large structural variants. Linked-read sequencing combines these strengths by using barcodes to tag DNA fragments from the same molecule. This keeps the low cost and high throughput of short reads while adding long-range information, making it easier to reconstruct haplotypes—something standard short-read data struggles to do. This approach has been used in many areas, including genome assembly and population genomics, but some early commercial platforms were discontinued. Newer methods have since been developed, including haplotagging, which is cheaper, simpler, and does not require specialized equipment. Through an ongoing collaboration with the Cornell Genomics Innovation Hub, we are expanding and improving on haplotagging linked-read chemistry.
Haplotagging, our choice for linked-read data
Haplotagging uses nanoscopic magnetic beads coated with DNA segments to bind to sample DNA and fragment long DNA. All of the newly created fragments will also be "tagged" with the same barcode, indicating that they originated from the same original DNA molecule. In the end, you get DNA fragments that are sized for sequencing on a short-read sequencer (like Illumina NovaSeq), and each of the DNA sequences will have a known association with other sequences that share the same barcode.
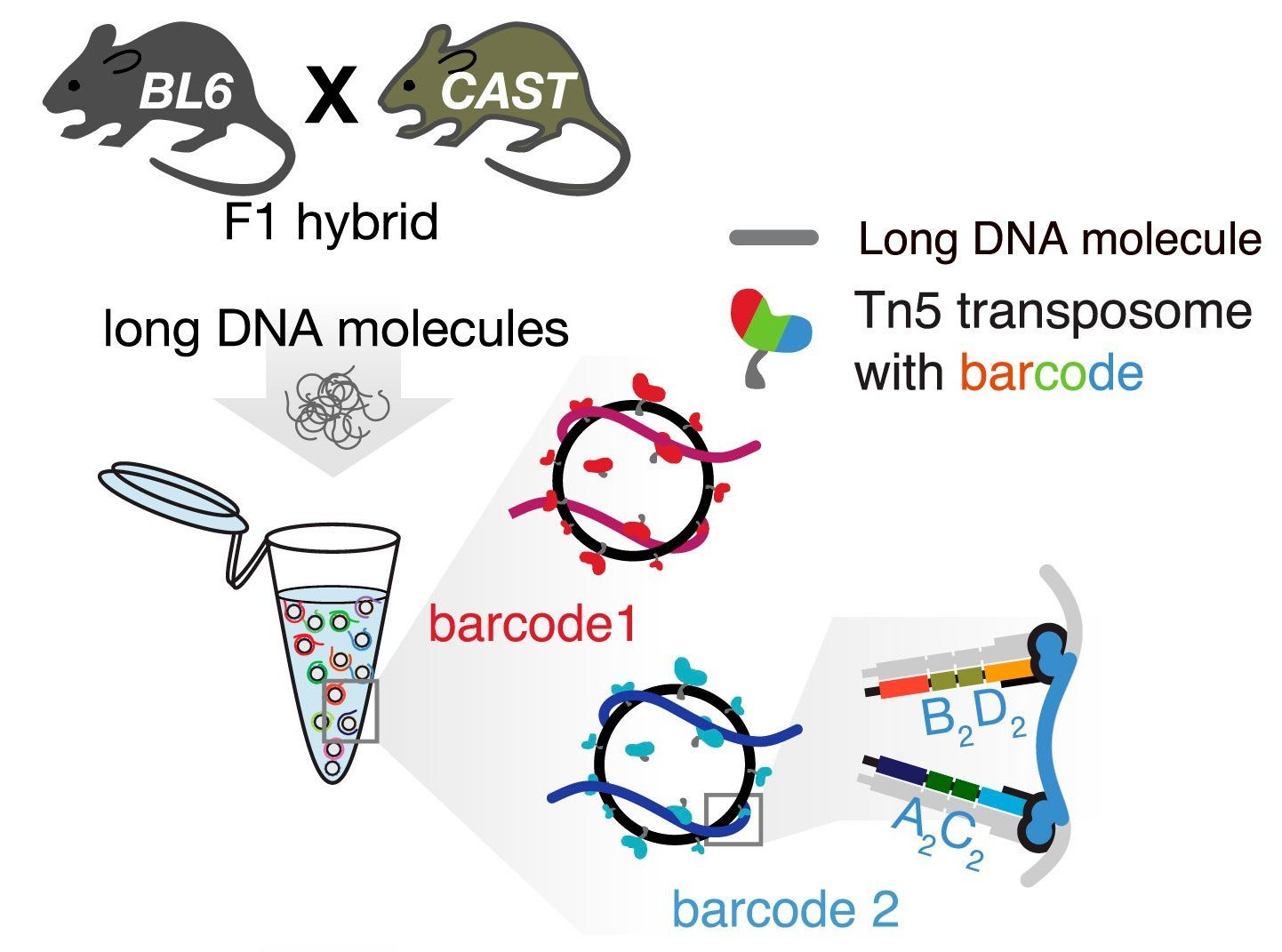 Haplotagging in a nutshell, from Meier et al. 2021
Haplotagging in a nutshell, from Meier et al. 2021
Creating supporting software
Despite the obvious improvements of linked-read sequencing over traditional WGS or reduced-representation methods, wider adoption of linked-reads depends on accessible data analysis tools. Earlier technologies benefited from user-friendly software (STACKS, ddRAD, etc.), but current linked-read approaches often rely on limited or platform-specific tools. To address this, Pavel wrote Harpy, a user-friendly pipeline that processes haplotagging data from raw sequences through alignment, variant detection, and haplotype reconstruction, with workflows designed specifically for linked-read data (although it has been modified to work on non linked-read data too).
 The Harpy logo
The Harpy logo
Using many of the previous software suites and tools as inspiration, Harpy is designed to be impressively user-friendly, which includes a considerable amount of detail in the command line interface, the descriptive error messages, the technical report system, and the software documentation. This effort has made using haplotagging (and linked-read data as a whole) nearly effortless for new users and experts alike.
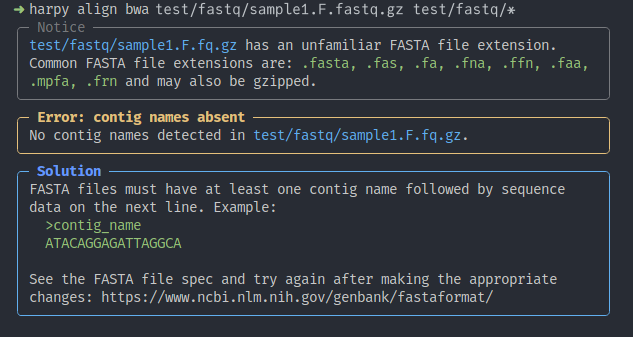 Example Harpy error message
Example Harpy error message
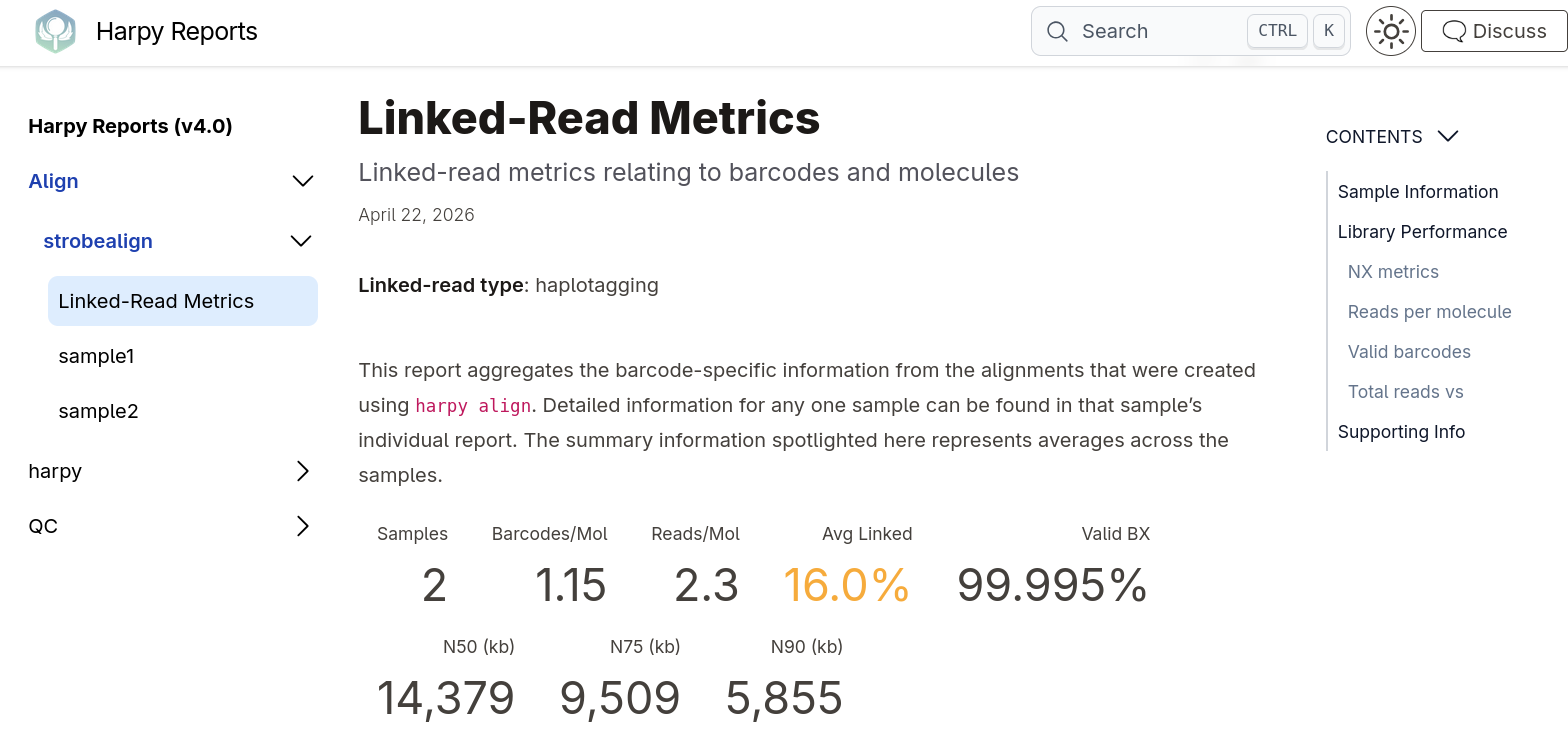 Example Harpy report
Example Harpy report
We use Harpy exhaustively, which also means that we can readily adapt it to new workflow patterns, cutting edge software/algorithms, and general usability improvements. In addition to Harpy, Pavel also wrote Mimick, a best-in-class linked-read data simulator, and Djinn, a multipurpose toolbox for manipulating and converting linked-read FASTQ and SAM data. You can tell that Pavel has a knack for naming linked-read software after dangerous fictional creatures. Be on the lookout for upcoming projects Wraith (structural variant caller) and Arachne (linked-read aware sequence aligner).